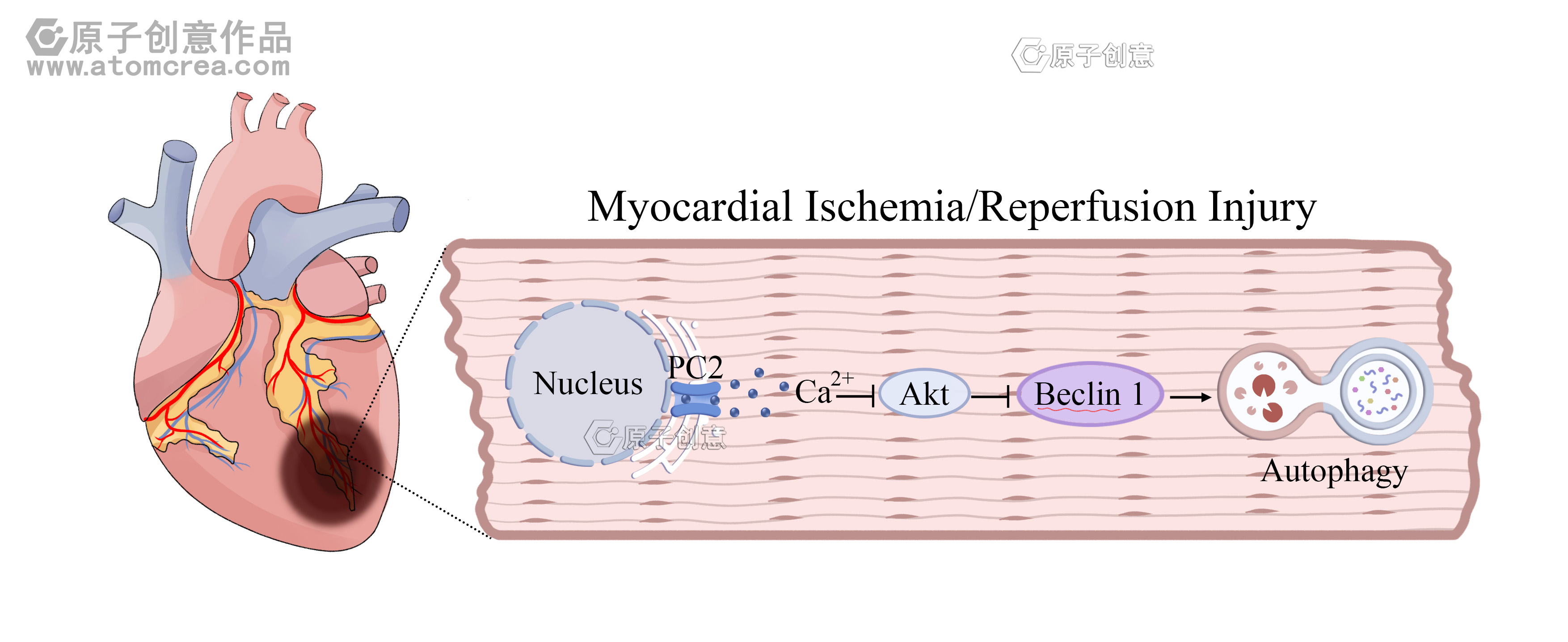

多囊蛋白-2的阻断通过Ca²⁺/Akt/Beclin 1通路抑制自噬,从而减轻心肌缺血/再灌注损伤
缺血性心脏病是发达国家和发展中国家的首要死亡原因。早期恢复缺血区域的血流可减轻缺血损伤。然而,再灌注可能会加重心肌损伤,这在全球范围内被称为心肌缺血/再灌注损伤(MIRI)[1]。已有充分研究表明,导致MIRI的病理生理机制与氧化应激、钙超载、细胞凋亡、内质网应激、自噬及细胞死亡相关[2]。许多研究指出,心肌缺血/再灌注(I/R)诱导的自噬是MIRI中心脏功能障碍的主要原因[[3], [4], [5]]。在缺血等特定条件下,自噬可降解功能异常的蛋白质并提供关键营养物质,从而抑制细胞凋亡和坏死。但再灌注期间过度的自噬可能会加重心脏损伤[6]。因此,在心肌I/R过程中,自噬的程度对其保护作用或损伤作用至关重要。
细胞外Ca²⁺内流和/或细胞内储存Ca²⁺释放的变化,会改变胞质内Ca²⁺水平,并成为自噬的关键调节因子[7,8]。Ca²⁺的复杂转运过程(包括内流和外流)由蛋白质通道控制[9]。越来越多的证据表明,多种阳离子通透性瞬时受体电位(TRP)通道会影响MIRI相关的生理系统,这表明它们可能成为治疗靶点[10,11]。多囊蛋白2(PC2)是TRP通道多囊蛋白家族的成员,由PKD2基因编码,其相关病理学特征已得到充分认识。PC2突变会导致常染色体显性多囊肾病(ADPKD),这是一种遗传性疾病,会导致双侧肾脏逐渐增大并最终引发肾衰竭[12]。PC2具有6个跨膜结构域,羧基末端含有Ca²⁺结合位点[13]。它是一种非特异性阳离子通道,参与维持细胞质内Ca²⁺的稳态,主要定位于内质网/肌浆网(ER/SR)[14]。值得注意的是,PC2最近被发现是一种常见的应激敏感蛋白,其表达会在细胞应激时升高[15]。事实上,在I/R损伤后,肾脏中PC2的表达显著增加[15]。此外,近期研究报道称,在饥饿状态下,自噬激活与心肌细胞中PC2介导的Ca²⁺超载有关,这表明在MIRI中,PC2可能通过调控细胞内Ca²⁺稳态而成为自噬的新型调节因子[16,17]。
服务对象 : 桂林医科大学
发表时间: 2025 年 2 月
发表期刊: Biochimica et Biophysica Acta (BBA) – Molecular Cell Research
文章链接: https://doi.org/10.1016/j.bbamcr.2024.119892







暂无评论内容